
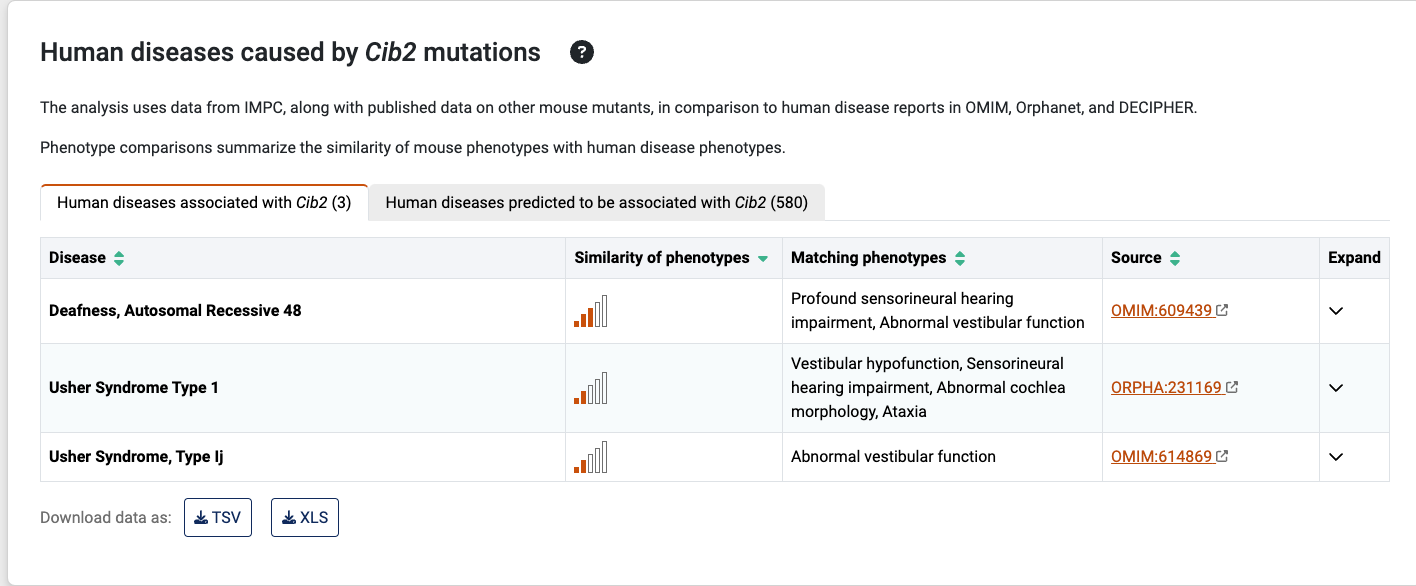
Disease associations
Once a mutant line has been phenotyped and phenotypes associated to it via the statistical analyses, its phenotypes are compared to those of patients. To do this, an algorithm called Phenodigm is used.
This procedure uses phenotype annotations:
- Mutant mice are encoded using terms in the Mammalian Phenotype Ontology
- Human patients are encoded using terms in the Human Phenotype Ontology, for all diseases described in OMIM, ORPHANET and DECIPHER
In brief, the Phenodigm algorithm proceeds in two stages:
- The first stage compares individual disease phenotypes (HP terms) to individual mouse phenotypes (MP terms). The outcome of each comparison depends on the semantic similarity of the terms and their prevalence. Thus, pairs of phenotypes that are biologically similar and relatively rare (e.g. human ‘Ataxia’ and mouse ‘ataxia’) obtain a high score, while pairs that refer to different biological entities and are quite vague (e.g. human ‘Abnormality of the cardiovascular system’ and mouse ‘adipose tissue phenotype’) obtain a very low score. Scores from this stage fall in the range [0, infinity], although typical values are [0, 6].
- The second stage of the calculation aggregates pairwise phenotypes scores into a single value. Conceptually, this aggregation is an attempt to summarize an overall similarity between the mouse and disease phenotypes, given the available data and annotations. The outcome of this stage is the Phenodigm score and falls in the range [0, 100], with low/high values indicating small/high concordance between the mouse and disease phenotypes.
Learn more:
Read the publication on Phenodigm: PhenoDigm: analyzing curated annotations to associate animal models with human diseases, Database 2013.