Institutional Members
Corporate Associates
Collaborations
Funding
Animal Welfare
About KOMP
Awards of Excellence
Training & Workshops
Workshops
Training
Getting Started with IMPC Data
IMPC Data Generation
How to Use Gene Pages
Citing IMPC Data
Allele design
IMPC Data Collections
Late Adult Data
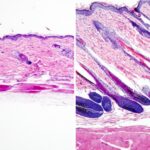
Histopathology
Essential genes
Embryo Development
Cardiovascular
Accessing the Data
Latest Data Release
Access via API
Access via FTP
Batch query
Advanced Tools
Disease Models
PhenoDCC Tools
GenTaR
OpenStats
IMPReSS
Embryo Viewer
Software
Latest IMPC Papers
Data Supporting IMPC Papers
Pain Sensitivity Associated Genes
Essential Genes - Linking to Disease
Essential Genes - Translating to Other Species
Sexual Dimorphism
Genes Critical for Hearing Identified
Genetic Basis for Metabolic Diseases
Papers Using IMPC Resources
The 100,000 Genomes Project is applying whole genome sequencing in a diagnostic setting to rare disease and cancer patients from…
Published: 16 Nov 2018
The entire genome of many species has now been sequenced, but the function of the majority of genes still remains…
Published: 28 Sep 2018
New research finds that scientists are still studying the same 10% of human genes and ignoring the rest, highlighting the…
Published: 19 Sep 2018
Researchers have discovered a new way to activate the stem cells in the hair follicle to make hair grow. The…
Published: 14 Aug 2018
Humans and mice share approximately 98% of genes, and have similar physiology and anatomy. This is because we share a…
Published: 9 Jul 2018
A recent study in the journal Nature Medicine utilises IMPC-generated mouse lines as a basis for their discovery. A summary…
Published: 22 Jun 2018
We are excited to be attending the European Society of Human Genetics (ESHG) 2018 conference this weekend in Milan! Visit stand…
Published: 11 Jun 2018
The International Mouse Phenotyping Consortium (IMPC) has been predominantly interested in using mouse models to understand human health and disease.…
Published: 25 May 2018
Leading international protein researchers have mapped two large unexplored parts of the human genome. Their work paves the way for…
Published: 15 May 2018
[embed]https://www.youtube.com/watch?v=4K4RDG1q6z8&t=97s[/embed] Brendan Doe talks about his work in using Cytoplasmic Microinjection to generate CRISPR mediated mutations. This method has recently…
Published: 20 Apr 2018







 19th August 20252026 Virtual IMPC Confere...
19th August 20252026 Virtual IMPC Confere... 22nd February 2019The development of a pote...
22nd February 2019The development of a pote... 5th February 2020IMPC Experts are Analysin...
5th February 2020IMPC Experts are Analysin...