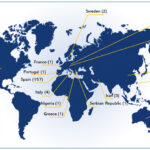
Institutional Members
Corporate Associates
Collaborations
Funding
Animal Welfare
About KOMP
COVID-19 Resources
Getting Started with IMPC Data
IMPC Data Generation
How to Use Gene Pages
Citing IMPC Data
Allele design
IMPC Data Collections
Late Adult Data
Histopathology
Essential genes
Embryo Development
Cardiovascular
Accessing the Data
Latest Data Release
Access via API
Access via FTP
Batch query
Advanced Tools
PhenoDCC Tools
GenTaR
OpenStats
IMPReSS
Embryo Viewer
Latest IMPC Papers
Data Supporting IMPC Papers
Pain Sensitivity Associated Genes
Essential Genes - Linking to Disease
Essential Genes - Translating to Other Species
Sexual Dimorphism
Genes Critical for Hearing Identified
Genetic Basis for Metabolic Diseases
Papers Using IMPC Resources
Please save the date for the 2nd virtual IMPC workshop in the 2024 series, titled ‘IMPC Data API Workshop' organised…
Published: 10 Apr 2024
We are happy to announce the first IMPC workshop for 2024 entitled ‘ Neurodegenerative consequences of genetic defects in taste…
Published: 12 Feb 2024
The Genetics Society of America is hosting a one-of-a-kind conference that offers highly relevant organism specific content as well as broader topic-driven…
Published: 19 Sep 2023
IMPC is hosting a Virtual Workshop organised by the Behavior and Sensory Working Group of the International Mouse Phenotyping Consortium…
Published: 30 Aug 2023
You are invited to attend the second International Mouse Phenotyping Consortium (IMPC) virtual open office hour on Wednesday 24th May…
Published: 19 May 2023
The PATHBIO - Morphological Mouse Phenotyping Course 2022 has been just completed in Barcelona, with the support of the IMPC.…
Published: 20 Jul 2022
A Scientific American documentary featuring Jacqui White of the Jackson laboratory (an IMPC member), was released last month. The film…
Published: 4 Jul 2022
Are you already an IMPC website user or a scientist interested in IMPC data? Have you read about IMPC but…
Published: 20 May 2022
2021 marked the 10th anniversary of the IMPC. Over the last 10 years the output and impact of the Consortium…
Published: 4 May 2022
On July 4th - 12th, 2022, the fourth course on Mouse Embryology, Anatomy, Histology, and Anatomical Basis of Imaging will…