

Jump to:
- What is Rare Disease?
- Why are Mice Important?
- Who We Are and What We Do
- How Has The IMPC Contributed to Rare Disease Research?
- Our Mouse Models for Rare Disease
- Rare Disease Day Events
What is Rare Disease?
So, what differentiates a rare disease from a more common disease? Definitions differ slightly depending on the country. A rare condition affects fewer than 200,000 people in the US (approximately 0.06%). In the EU, a rare disease affects 1 in 2,000 (0.05%).
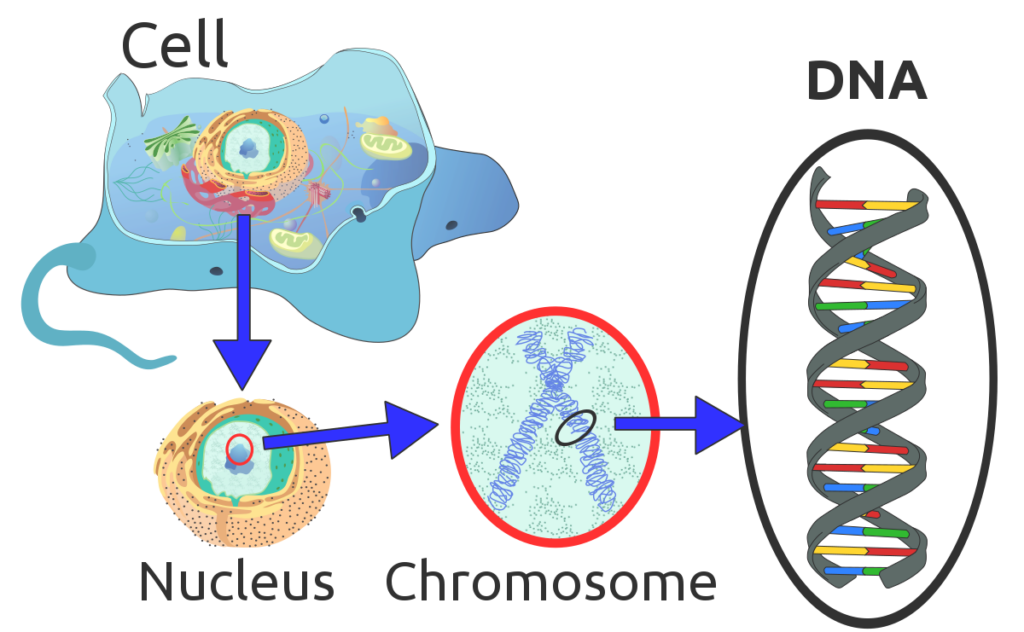
Rare diseases are caused by many things, but the majority are thought to be genetic – caused by changes in our DNA. In humans, DNA exists in every cell as two long chains twisted in a spiral shape called the double helix. The two chains connect like a ladder, with the rungs being pairs of four possible shapes, called nucleotides. Nucleotides include the pairs A and T or G and C. Genes are distinct stretches of the double helix. The nucleotide letters form a code in each gene segment that is used to make proteins.
DNA is actually quite long. In each cell, you have 6 feet of DNA if it were stretched flat out of its helix shape. If you stretched out all of the DNA in a single human, it would be long enough for about 150,000 round trips to the moon.
Even in a single cell, 6 feet of DNA is a lot to contain, so the double helix length is compacted down and scrunched up into tight ‘X’ shapes called chromosomes. We have 23 pairs of chromosomes in each cell or 46 in total.
Sponk, Tryphon, Magnus Manske, User:Dietzel65, LadyofHats (Mariana Ruiz), Radio89,, CC BY 3.0, via Wikimedia Commons
{kind=link}
A genetic disease can happen when genes or chromosomes change. Sometimes genes mutate during the copying process needed for cell division. When this happens randomly in an egg or sperm cell or very early on during embryo development, it’s called a ‘de novo’ mutation. De novo mutations can cause very rare diseases in individuals with no family history of disease. Gene mutations can, however, also be passed down through families, causing inherited disease.
Chromosomal abnormalities can also cause genetic disease. For example, part of a chromosome can be accidentally deleted during cell division or accidentally moved or flipped. A much larger section, such as several genes, could be lost.
Why do gene or chromosome mutations cause genetic disease? If the code in a gene is changed, the protein that the gene codes for may no longer work. Proteins play a vital role in all processes in the human body. They allow cells to talk to each other, oxygen to travel around the body, combat against infectious disease, correct growth, nutrient absorption, thought, movement, and much more.
When proteins no longer work properly, these vital processes become disrupted or stop entirely, causing disease.
Why are mice important?
Mice are a vital resource in research. They share about 97% of our genes, have similar cells, organs and systems, are easy to maintain and breed, and are widely accessible for scientists. Research using mice has contributed massively to our understanding of human biology and the development of fundamental medical and scientific methods, tools and treatments.
Researchers tested penicillin in mice with streptococcal infections. The tests proved that penicillin killed bacteria in live organisms and allowed researchers to find safe dose levels for humans, allowing its wide use in the modern-day.
Other historical contributions of mice include the vaccine against Haemophilus influenza (a cause of meningitis), organ transplant research, and early testing of tamoxifen (a breast cancer treatment.) This list is growing larger every year, particularly in the realm of human genetic disease.
Who We Are and What We Do
The International Mouse Phenotyping Consortium, IMPC, comprises 21 of the leading mouse genetics centres worldwide. We produce a type of mutant mouse called knockout (KO) mice, where we ‘turn off’ a single gene so that it no longer works at all. We then ‘phenotype’ the knockout mouse by conducting tests and activities. The data collected is compared to a strong set of data from normal mice to find any differences. If any significant differences are found, we list them in our database as a ‘phenotype’. This process is called ‘phenotyping’. We aim to make a KO mouse for every gene that codes for a protein.

By doing this, we have been making a massive catalogue of what functions a gene might have and how it might cause disease if the gene stops working. This information can be applied to humans. It can help us find new genes that may cause disease and possible gene targets for treatments. Our open-access database is a great resource for scientists that may be interested in researching an unexplored gene. By creating the KO mice and freezing and storing their sperm cells, we’re also reducing the number of mice needed to re-produce a mouse type.
KO mice can either be ‘homozygous’ or ‘heterozygous’. We have two copies of every gene – one from our mother and one from our father. Usually, there are several variants of a gene within a population, each with a slightly different code. Simply, ‘homozygous’ is when you have two copies that are identical and ‘heterozygous’ is when the copies are different. Zygosity and dominance are important in both common and rare diseases, as well as normal biological functions. Dominance involves whether gene variants are dominant or recessive. A dominant variant of a gene can overpower the effect of a recessive variant. Depending on how the two variants interact, the strength of dominance can vary.
In disease, there are some good examples of zygosity and dominance playing a role in inheritance and disease severity. Marfan’s syndrome, for example, only needs one disease variant to cause disease. This means an individual with Marfan’s syndrome is usually “heterozygous dominant” because they have two different copies, one of which is dominant.
Sickle cell disease is another great example. Here, individuals can be carriers (sickle cell trait) or full sufferers (sickle cell disease.) Those with the full disease have two identical disease gene variants, so they are homozygous. These variants are actually recessive to a healthy gene variant. Carriers would be heterozygous recessive and can have symptoms when exercising or dehydrated.
We make homozygous mouse models, where we knockout both gene copies, and heterozygous ones, where we knock out only one copy. Many diseases, rare and common, are associated with age so, at the IMPC, we phenotype groups of our mice at different ages. We, therefore, classify phenotypes as ‘embryo’, ‘early’, ‘middle’ or ‘late’ depending on when the phenotyping was conducted.
IMPC mice are available for all researchers to use in rare disease studies. Our data is also free to access and use.
In fall 2021, the IMPC organised its 10-year celebratory conference. Session VIII of the conference included presentations and discussion on the wider translation of mouse genetics on genomic and precision medicine including the utility of bringing diverse mouse resources to bear for the analysis of disease systems, including rare disease systems. View the on demand video from our conference.

How Has The IMPC Contributed to Rare Disease Research?
Since 2021, the IMPC has established a collaboration with Solve-RD project and through their dedicated platform RDMM –Rare Diseases Models and Mechanisms Europe, different labs from partner institutes of the IMPC were selected to provide mouse models for different genes identified in rare diseases.
- IMPC researchers conducted human and mouse essentiality screens. In this study, they created a new categorisation system to class genes by how essential they were. Essential genes are very important; they usually ensure that an organism can develop and grow or allow individual cells to function well. These genes are so important that if they stop working due to mutation it can cause the organism to die before or shortly after birth. The researchers found that essential mouse genes were more likely to be associated with human disease. The new system makes it easier to find ‘novel candidate genes’ for rare human developmental disorders, which can be difficult to diagnose when caused by an unknown gene. Read our article here for more.
- Our IMPC consortium member CAM-SU GRC developed a new algorithm to find candidate genes involved in circadian rhythms. These rhythms are our daily processes and cycles, such as sleeping and eating. They can be disrupted by internal and external factors, like jet-lag or shift work. When chronic disruption, or ‘misalignment’, occurs due to genetic mutations, it can cause sleeping, eating or psychiatric disorders. The algorithm can search genes with existing mouse model data to find ones associated with misalignment. This helps researchers more easily find novel candidate genes involved in metabolic, sleep and eating disorders. Read our article here for more.
- Our IMPC consortium member CNR Monterotondo tested a custom-made antibody this year. This specially-made antibody targets ‘connexin (Cx) hemichannels’, proteins on the surface of human cells that allow substances to enter and exit. Cx Hemichannels are important for cell-to-cell communication and play a role in many vital biological processes. The study focused on a single type of Cx channel called Cx30. A mutation in the Cx30 gene causes the channel to become ‘leaky’, which causes a rare disease called Clouston syndrome (abnormal development of hair, nails, skin, teeth and sweat glands.) The researchers found that the antibody successfully blocked leaky Cx30 channels, meaning it could potentially alleviate symptoms. It also opens the door for more antibodies targeting other leaky Cx channels, such as Cx32 in Charcot-Marie-Tooth disease. Read here for more.
The IMPC also helps target genetic variants of unknown significance (VUS) via KOMP2, which is part of the IMPC. KOMP2 produces mouse models used to provide clinically useful data to determine the pathogenicity of VUS, accelerate diagnosis, and inform clinical decision-making and patient care. In 2 years, KOMP2 has generated 20 mouse models for patient-specific genomic variants. Targeted phenotyping allows us to determine the extent to which the genomic variant is likely causative of, rather than merely associated with, the patient’s disease or syndrome.
For example, KOMP2 produced a homozygous KO model of the Tbc11d5 gene. A single case study of a female child with a heterozygous mutation in TBC1D5 showed symptoms of developmental delay, infantile spasms, progressive hearing loss in both ears and increased blood pressure.
The KO mouse showed phenotypes of impaired hearing function, reduced sociability, cognitive impairment and anxious/compulsive behaviour.
Our Mouse Models for Rare Disease
Atp1a3 and Alternating Hemiplegia of Childhood (AHC) 2
AHC 2 is caused by a heterozygous dominant mutation in the ATP1A3 gene. It’s characterised by a range of nervous system and developmental issues, such as paralysis, developmental delay, abnormal posture and repetitive movements.

Patient groups for AHC include the Alternating Hemiplegia of Childhood Foundation, AHC Association of Iceland, ENRAH, Cure AHC and AHC International Alliance, among others.
.
Abcg8 and Sitosterolemia 1 (STSL1)
STSL1 is caused by a homozygous or compound heterozygous recessive mutation in the ABCG8 gene. It’s characterised by uncontrolled absorption of cholesterol and cholesterol-like molecules in the gut from food. The abnormally high intake of cholesterol causes the build-up of fatty plaques in arteries, leading to coronary heart disease and heart attacks. Other symptoms include joint pain, arthritis and blood cell abnormalities, like enlarged platelets and too many red blood cells.
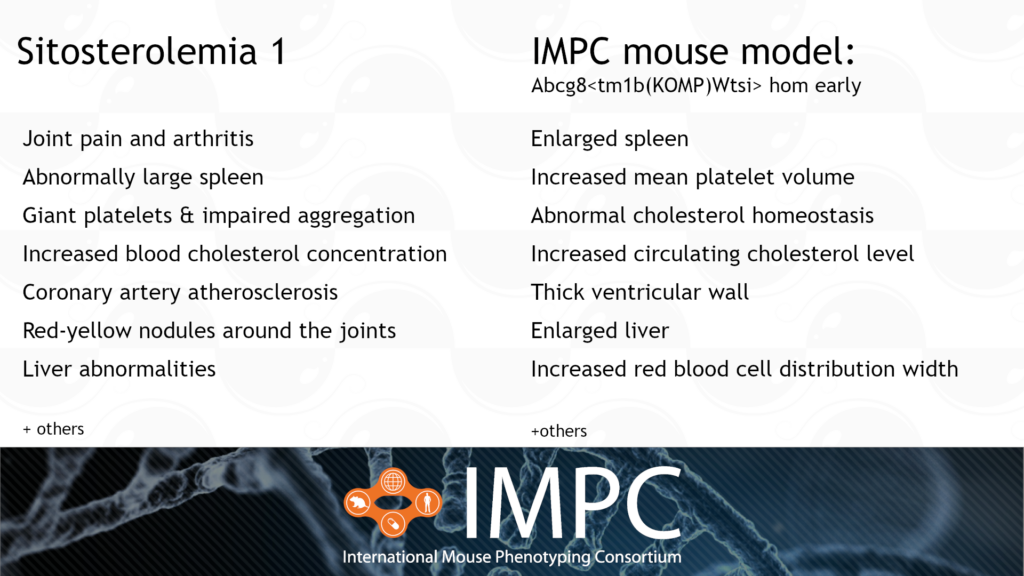
Patient groups for STSL1 include the Sitosterolemia Foundation and, Heart UK and other broader heart disease organisations, such as the American Heart Association.
.
Fmo3 and Trimethylaminuria (TMAU) or “Fish-Odour syndrome”
TMAU is caused by a homozygous or compound heterozygous recessive mutation in the FMO3 gene. It’s characterised by large amounts of trimethylamine in the body, which is then excreted in the urine, sweat and breath, causing a fishy odour. Our IMPC late-adult mouse model is the first model to capture spleen phenotypes relevant to TMAU.

Patient groups for TMAU include MEBO Research and Metabolic Support UK.
.
Tent5a and Osteogenesis Imperfecta Type XVIII (OI18)
OI18 is caused by a homozygous recessive mutation in the TENT5A gene. It’s characterised by bowing of the long bones, wormian bones, multiple fractures and vertebral collapse, all from birth or early life.

Patient groups for OI18 include the Canadian Osteogenesis Imperfecta Society (COIS), Children’s Brittle Bone Foundation, Hypermobility Syndromes Association, Osteogenesis Imperfecta Foundation and the Brittle Bone Society.
.
Polg2 and Progressive External Ophthalmoplegia with Mitochondrial DNA Deletions 4 (PEOA4)
PEOA4 is caused by a heterozygous dominant mutation in the POLG2 gene. PEOA4 is a mitochondrial disease, meaning it affects a small part of every cell called the mitochondria. The mitochondria is a vital part of the cell because it’s where energy is produced. This energy is the currency all cells use to perform different tasks and processes. Mitochondria are also a unique part of the cell because they have their own DNA, separate from our double helix DNA. POLG-related disorders like PEOA4 are caused because a protein that helps replicate mitochondrial DNA is disrupted. PEOA4 is characterised by abnormalities in skeletal muscle, the nervous system, the liver and the gastrointestinal tract.
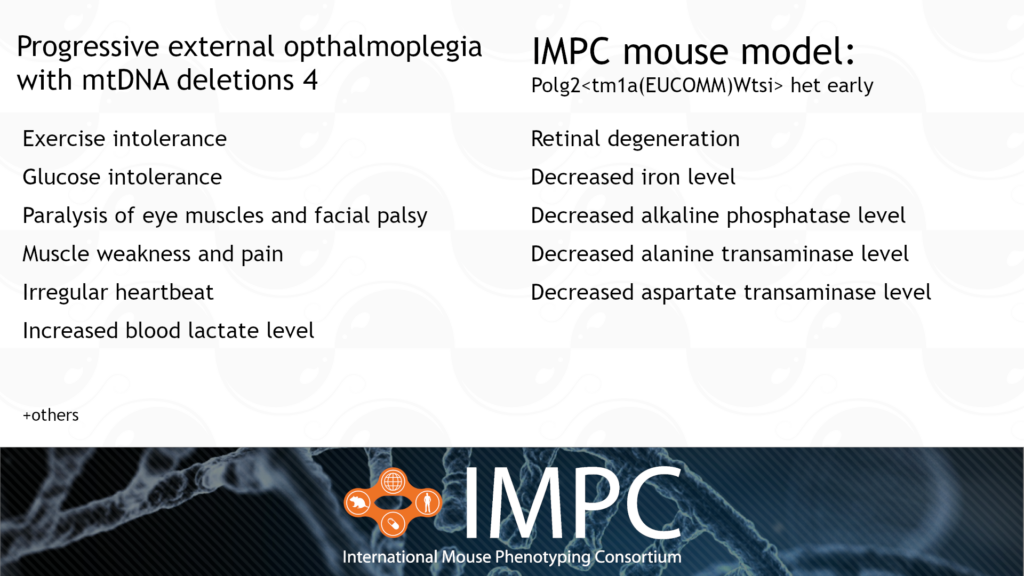
Patient groups for PEOA4 include the United Mitochondrial Disease Foundation, Mitoaction and the Mitochondria Research and Medicine Society.
We’re producing and phenotyping many more genes related to rare diseases. Do you have a gene of interest? Use our search function at the top of this page to see if we have models and data for that gene. Check out Rare Disease Day 2022 on their website and Twitter page!