Welcome to our Help Pages!
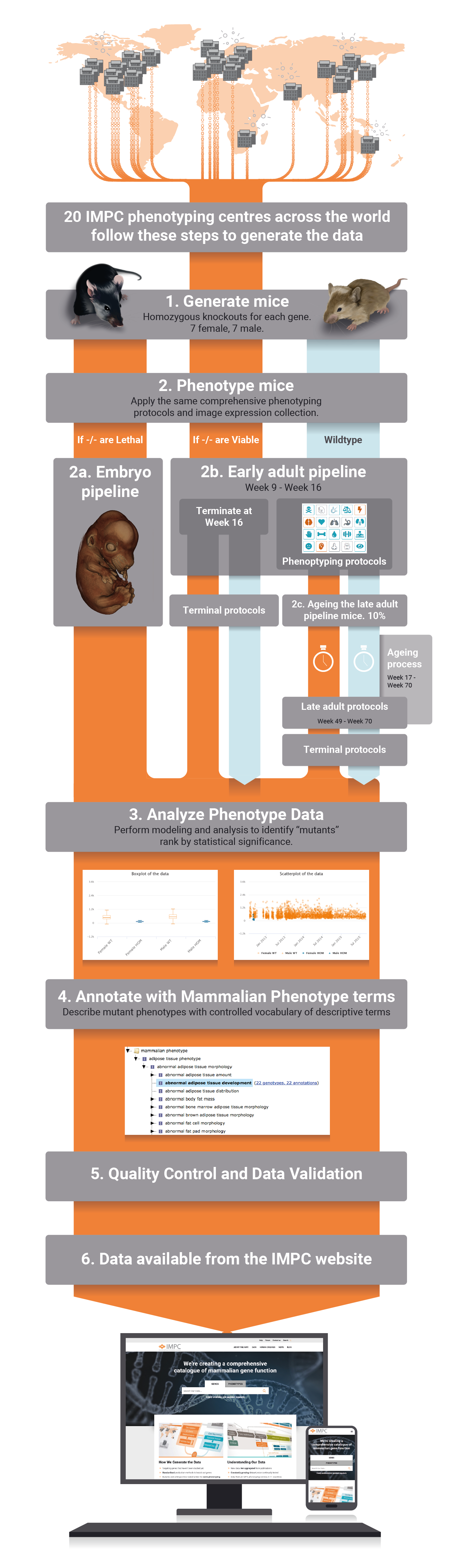
The International Mouse Phenotyping Consortium (IMPC) is an international research infrastructure which aims to identify the function of every protein-coding gene by creating and phenotyping single-gene mouse knockout lines for each of the 20,000 or so protein-coding genes in the genome. Our database is continuously growing as new data is being generated. IMPC data is allowing the generation of new hypotheses that are increasing our understanding of the mammalian genome, forming the basis of new research, and identifying disease associations. IMPC papers exemplify the advances and contribution to the generation of new knowledge that has been possible with the emergence and analysis of IMPC data.
What information can I find in this website?
All phenotype data and images of all knocked out mouse generated by the IMPC are accessible through our website, organised by gene or phenotype. Deviations from wildtype phenotypes on single gene knocked out mice allows to identify the physiological system that is disrupted when a gene is disabled. Thus, these data effectively allows to generate gene-phenotype associations. The IMPC has developed specific in-house protocols for high-throughput mouse production and tracking, as well as standardized mouse phenotyping and data analysis pipelines, which can all be accessed through this website.
How can I access the data?
Several entry ways are provided. In the website search bar, which is available on the home page and all pages, you can specify a gene or phenotype of interest. Alternatively, you can browse our bespoke data collections or the scientific papers. For specific reports or bulk downloads, both Programmatic and Non Programmatic Data Access are supported. All data and images are available to use, free of charge! The IMPC dataset is publicly available under the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
How can I learn more?
We invite you to learn more about the IMPC through these pages. You may want to visit our Getting Started with IMPC data section. We have a dedicated webinar and online training course. Our webinar introduces IMPC findings as well as the phenotyping pipelines, statistical analyses, and engagement with clinical and external consortia. Our online training course exemplifies IMPC website navigation and data accessing, with numerous website screenshots, interactive content and exercises. A brief description of the project can be found here, and consortium publications are listed here. The IMPC blog provides information on recent relevant publications, interviews and news.